The mechanism of the spontaneous detonation of ammonium nitrate inreactive grounds
1. Introduction
Ammonium nitrate, or AN (NH4NO3), is the basis of most explosivesused in the mining industry. Its detonation usually must be deliberatelyinitiated, but there have been a number of instances when it hasspontaneously exploded, at great risk to life and property. One of thecauses has been identified as the presence of pyrite (FeIIS2) in contactwith the explosive. Another is the presence of naturally high tempera-tures in the ore body, from some geothermal source. These causes aretermed‘reactive ground’and‘hot ground’, respectively[1].The explosives industry has developed emulsion explosives due totheir superior water-resistance, and low environmental impact[2,3]over traditional explosives. However, when sulfide ores are present, orin hot grounds, inhibited emulsion explosives must be used to preventpremature detonation of the explosive. The explosives industry achievethis by the addition of urea, to extend the‘sleep time’of the product-the quiescent time between contact with the reactive ground and theexothermic process that leads to detonation[4,5]. The role of urea as aninhibitor was the original subject of this research. The work led to anew, alternative understanding of the mechanism of the spontaneous,and of the inhibited, decomposition reactions.
1.1. Chemistry background
Ammonium nitrate decomposes in an exothermic reaction(ΔH=–36 kJ mol−1) to produce three moles of gaseous products foreach mole of solid reactant:→+NHNONO2HOsg43()2()2(g)(1)The reaction can be made more exothermic (ΔH=–280 kJ mol−1),with more gaseous products, if some oxidisable fuel is added:+→ ++2NH NOC2N4H OCOsgg43()2()2(g)2()(2)Hence, the standard ammonium nitrate explosive mixture is termedANFO, for an“ammonium nitrate fuel oil”mixture.The decomposition temperature of pure ammonium nitrate is170 °C, but recently Gunawan and Zhang[6] calculated that an in-timate mixture of ammonium nitrate and pyrite can decompose attemperatures as low as 50 °C in blast holes more than 0.2 m in dia-meter. This is consistent with manyfield observations of detonations atlow ambient temperatures. We have analysed the heatflow in thesesystems elsewhere[7].The same initial reactions occur in acid mine drainage, which has been extensively studied[8,9]. Parallels can be made between the twoprocesses and analogies usefully drawn. Water is required in both cases,implying that soluble species are involved. Thefirst step in the processis the oxidation of pyrite by air. The oxidation product of the sulfurcould be various substances, such as SO2,SO3, thiosulfate, etc. For il-lustrative purposes we choose SO2, because it is detected as a product inreactive ground environments; however, this choice does not affect theconclusions of the argument. For example, oxygen from the air oxidizesthe disulfide anion to SO2 The process now becomes autocatalytic, as more acid is producedand more Fe(III) dissolves. The rate-limiting step in this inorganic cyclethen becomes the oxidation of Fe(II) to Fe(III) by oxygen, but in thefield this is accomplished rapidly by bacteria. In mine sites wherebacteria are present, pH values can range from 0.7–3.0 [12]and ferric(Fe(III)) concentrations from 1 to 20 g/L[13,14]. Such conditions havebeen shown to lower the thermal decomposition of ammonium nitrate[15,16].The thermal profile of the decomposition process comprises threestages: an induction period, an intermediate stage and thefinal highlyexothermic decomposition. (Fig. 1) The reactions described abovecould explain the observation of the induction period in the thermaldecomposition of ammonium nitrate explosives caused by reactiveground. Some preliminary studies have indicated an inverse correlationbetween initial acidity and the induction time. According to Rumball,acid accelerates the rate of the initial stage and has little or no effect onthe intermediate stage[17]. 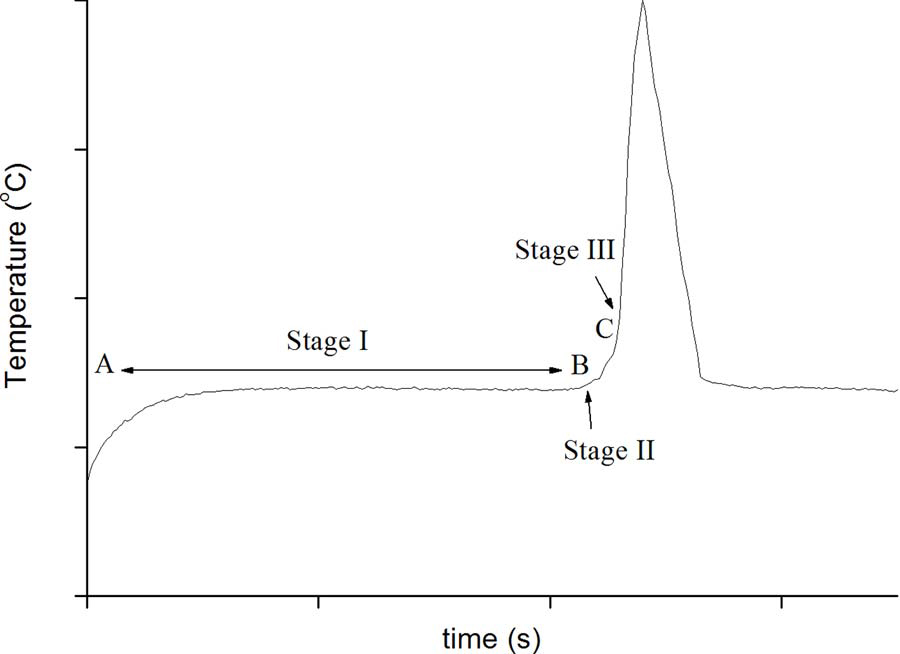
The initial stage of the process isinterpreted as the slow reduction in pH until the rapid and exothermicoxidation by Fe(III) accelerates.The preferred method of controlling both acid mine drainage andreactive ground has been to maintain a high pH through the use ofalkaline substances. The use of solid bases, such as limestone, is noteffective, however, as the Fe(III) precipitates on the surface and passi-vates the remaining solid base, a process termed‘armoring’, renderingit ineffective.Accordingly, the use of urea is preferred, which homogeneouslygenerates the weak bases ammonia and carbonate by hydrolysis, andhence consumes protons (Eq.(8))++ → +++CO(NH )2H2H O2NHH CO222423(8)There is compelling empirical evidence in the industry that urea isan effective inhibitor of the thermal decomposition of AN in reactiveground. The mechanism of this inhibition is uncertain. The hydrolysisof urea is known to be a slow reaction, which proceeds at a rate that isindependent of pH[18]. The length of the induction period could belimited by the total consumption of the urea. Alternatively, if the rate ofacid generation is greater than the rate of urea hydrolysis, then the pHof the system could slowly drop, despite the partial neutralisation bythe urea hydrolysis, until it reaches an acidic condition that allows anautocatalytic runaway decomposition. Finally, the urea could act as aninhibitor by a mechanism not involving its acid-base chemistry. Thisthird alternative is in fact our preferred explanation from the evidencewe have accumulated.2.
2 Experimental
2.1. Materials
Several sources of pyrite were used. RG1 was supplied by DynoNobel and is a reactive grade ground sample containing∼2.50% byweight of adsorbed water, and a pyrite content of less than 10 wt%. Theremaining material is a mixture of clays, quartz and organic matter. Theparticle size is less than 50μm on average. Pure pyrite (PY) was ob-tained from Spectrum Chemicals and is 100% oxidized pyrite with agrain size of 200–400μm.Ammonium nitrate, AN, (Acros Organics, 99 +%) was used as re-ceived but was ground in a mortar prior to use to break up any largeclumps. Dodecane (Sigma,≥99%), iron (II) sulfate 7 hydrate(BDH, > 99.5%), iron (III) sulfate 5 hydrate (Fluka), urea (Ajax che-micals, 99.5%), hydrazinium sulfate (Ajax chemicals, > 99.5%), zincoxide (Aldrich, 99.9%, < 1μm), kaolin (Kaolin Australia, Pty Ltd,Eckafine BDF), hydrotalcite (Sigma) and Basolite C300 (BASF) wereused as received. Sodium nitrite (Mallinckrodt), diacetyl monoxime,DCM (Fluka), thiosemicarbazide, TSC (BDH), phosphoric acid (85%,Ajax Finechem Pty. Ltd), sulfuric acid (96%, Ajax Finechem Pty. Ltd),iron (III) chloride 6 hydrate (Merck), and PIBSA-DEEA (Clariant) werealso used as received.
2.2. Urea determination
Urea was determined by UV–vis spectroscopy at a wavelength of525 nm using diacetly monoxime, DCM and thiosemicarbazide, TSC[19]. An acidic ferric solution was made containing phosphoric acid(100 ml), sulfuric acid (300 ml), water (600 ml) and ferric chloride(0.10 g). DCM and TSC were mixed (0.50:0.01 g) and made to volume(100 ml). When ready to use, the chromogenic reagent containing theacid solution (2 parts) and DCM/TSC solution (1 part) were mixed. Ureastock solutions were prepared containing∼20 ppm urea.Standard urea solutions were prepared by diluting stock urea solu-tions in water. The urea solution (0.32 ml) was mixed with the chro-mogenic solution to 10 ml, covered with aluminum foil and heated inboiling water for 10 min. The sample was cooled rapidly in ice and theUV–vis spectrum was measured from 400 to 600 nm.
Six samples containing 5 wt% urea (based on AN), ammonium ni-trate (0.9 g), reactive ground 1 (0.9 g) and weathering solution con-taining dissolved urea (0.047 g) were prepared and placed in twothermosflasks and heated to 55 °C in a sand bath. Samples were re-moved at selected time intervals, thefirst afterfive minutes and the lastafter 20 days. The samples were quenched with water (8.4 g) and thepH measured. More water was added (30 g total) and the slurry wasthenfiltered through a 0.2μmfilter in a 50 ml volumetricflask con-taining a drop of concentrated sulfuric acid. The solution was furtherdiluted (1.0 ml into 50 ml) and 0.32 ml was dispensed into 10 ml vo-lumetricflasks to which the chromogenic solution was added to vo-lume. The sample was heated, cooled and then the UV–vis spectrumwas measured from 400 to 600 nm. Inhibition studies involved addinginhibitors to the PY/AN/WS mixture, sealing and capping with a syr-inge, then heating to 55 °C in a glycerol bath. The reaction was notedwhen NOxbegan to be generated in the syringe.
2.3. Weathering solution
Synthetic weathering solution was freshly made, containing iron (II)sulfate 7 hydrate (0.245 g), iron (III) sulfate 5 hydrate (0.50 g), andwater (3.3 g). The mixture was gently sonicated until fully dissolved.
2.4. pH measurements
Mixtures of reactive ground (RG1), ammonium nitrate (AN), andweathering solution (WS) were reacted (0.9:0.9:0.2 g) in small glassvials and heated in a water bath at 55 °C or 80 °C forfive minutes. Afterthis time the sample was quenched with∼6.5 g water and the pHmeasured.
3. Results and discussion
3.1. Tests of the acid neutralisation hypothesis
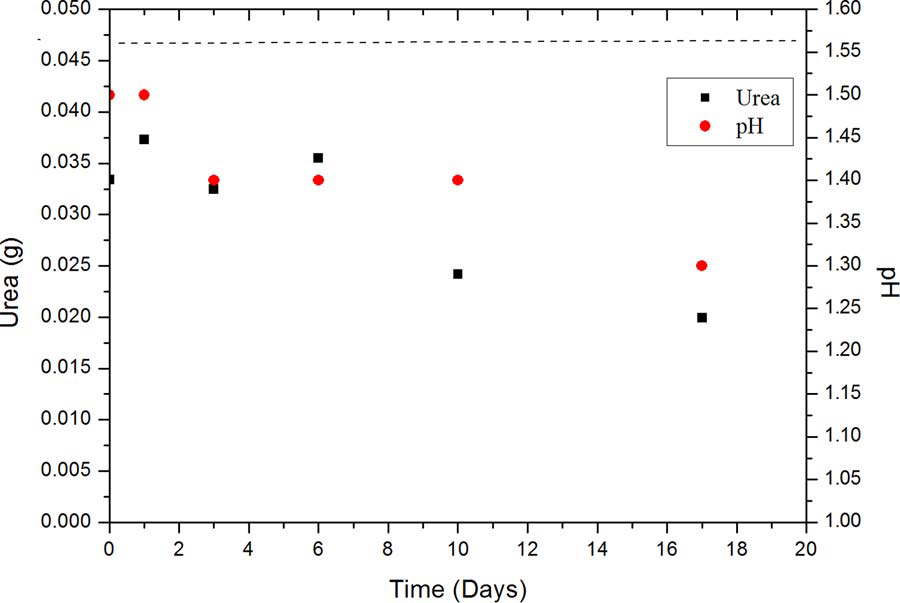
Reactive ground and pure pyrite was used and characterized by XRD(Fig. 2). The reactive ground sample contained mixtures of mineralsconsisting predominantly of quartz (Q), with some clinochlore (C), aswell as some pyrite mineral (P)[20]. The spectrum pyrite was 100%pyrite. Six reactions containing ammonium nitrate (AN), reactiveground (RG 1) and weathering solution (WS), with 5 wt% urea, wereprepared and sampled after 5 min, 1, 3, 6, 10 and 17 days. Afterquenching the samples with water the pH was measured and the totalurea analysed by UV–vis. After 17 days of reaction, the consumption ofurea was only partial; the urea decreased from an initial mass of 0.046 gto∼0.02 g. During this time, the pH of the slurry decreased from 1.5 to1.3 ( Fig. 3). If urea were hydrolysing to produce base, then the pHshould be greater than 1.3 after this time. Similar results were found atlower urea concentrations; with 0.2 wt% the urea deceased by one-thirdprior to the decomposition process at the end of the induction period.We conclude that the hydrolysis of urea is too slow to neutralise acidsignificantly, especially during the early stages (< 1 day) of the reac-tion, and that the presence of excess urea does not increase the hy-drolysis rate. This is consistent with literature reports that the hydro-lysis reaction is slow, with a rate constant of 8.4 × 10−10s−1at 25 °C[21].